

ריצוף ברמת התא הבודד
ריצוף ברמת התא הבודד בוחן את רצף הנוקלאוטידים מתאים בודדים תוך שימוש בטכנולוגיות ריצוף הדור הבא ( NGS, Next Generation Sequencing), טכנולוגיות המאפשרות הבחנה בדקויות ההבדלים בין תאים בודדים שונים בתוך רקמה ולאפיין תתי אוכלוסיות פונקציונאליות של תאים המאפשרת הבנה טובה יותר של התפקוד של תא בודד בהקשר של מיקרו-סביבה שלה.[1]
רקע
התא הוא היחידה הבסיסית של החיים. החומר התורשתי בתא האנושי נמצא בגרעין וארוז ב- דנ"א ומורכב מכ-3.3 מיליארד זוגות של בסיסים - נוקלאוטידים ומ-600 מיליון בסיסים של mRNA. לפי הדוגמה המרכזית של הביולוגיה המולקולרית המידע התורשתי משועתק מדנ"א לרנ"א ואז מתורגם לחלבון. כמויות אלו של נוקלאוטידים יקרות לריצוף בשיטות הישנות של ריצוף סנגר.
שיטות NGS (ריצוף הדור הבא) משמשות לריצוף ברמת התא הבודד ומורכבות מהשלבים הבאים:
- שבירת הרקמה הביולוגית לתאים בודדים
- שבירת התא ומיצוי החומר הביולוגי (דנ"א או רנ"א לפי הצורך).
- הכנת ספרייה - הכנסת מקטעים ברצפים ידועים לצורך זיהוי
- הגברה באמצעות PCR.
- ריצוף
- ניתוח ביואינפורמטי של התוצאות.
בניתוח ברמת התא הבודד כמויות החומר הביולוגי ההתחלתי מכל תא הן בסדרי גודל של פיקוגרמים ולכן נדרשת הגברה גבוהה מה שגורם לרעש גדול בתוצאות. ניתוח ברמת התא הבודד מאפשר ללמוד על ההטרוגניות של רקמה ביולוגית ועל תתי האוכלוסיות המרכיבות רקמה הנראית מבחוץ כאחידה וכן לזהות תתי אוכלוסיות כולל תתי אוכלוסיות נדירות כאוכלוסיית תאי אב סרטניים. ניתן ללמוד על תפקודים תאיים בהרחבה.
ריצוף ברמת התא הבודד התפתח לאחרונה בשל מספר חידושים והוא מאפשר לחקור בעיות כגון: הטרוגניות של רקמות, אוכלוסיות נדירות של תאים, קשר בין אוכלוסיות של תאים ורקמות מוזאיקה.
ריצוף ברמת התא הבודד נבחר על ידי קבוצת Nature לשיטה הנבחרת לשנת 2013.
ריצוף דנ"א ברמת תא בודד
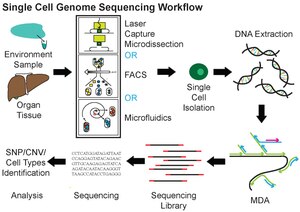
ריצוף ברמת התא הבודד מערב הפרדה לתאים בודדים, ביצוע הגברה לכל הגנום (WGA), בניית ספריות לריצוף המתאימות לטכנולוגיית הריצוף שבשימוש ולבסוף ריצוף הספרייה שנתקבלה במערכות ריצוף הדור הבא NGS (למשל מתוצרת אילומינה או איון-טורנט). ניתן להשתמש בריצוף ברמת התא הבודד במחקרי מטהגנומיקה (Metagenomics) ובריצוף אורגניזמים חדשים. ניתן לשלב ריצוף ברמת התא הבודד עם שיטות למיון תאים בתפוקה גבוהה של מיקרואורגניזמים ושל סרטן. בריצוף ברמת התא הבודד נפוצה שיטת הגברה שאינה PCR הנקראת הגברה בריבוי העתקות (Multiple Displacement Amplification), אשר מאפשרת מחקר בגנטיקה של חיידקים, אקולוגיה ומחלות מידבקות. נוסף לכך, מידע שהופק ממיקרואורגניזמים יכול לבסס תהליך של גידולם בתרבית בעתיד. כמה כלים שיכולים לשמש עבור ריצוף גנום ברמת תא בודד כוללים: SPAdes, IDBA-UD, ואת Cortex ו- HyDA.
שיטות הגברה
הגברה בשיטת ריבוי העתקות (Multiple displacement amplification) מאפשרת הגברה של חומר גנטי מרמות של פמטו-גרמים בודדים למיקרוגרמים לצורך שימוש בתהליכי ריצוף. החומרים הנדרשים לביצוע הריאקציה הזו הם פריימרים אקראיים ואנזים DNA פולימראז המופק מבקטריופאג' phi29. בשונה מריאקציית PCR להגברה, הריאקצית MDS מתרחשת בטמפרטורה קבועה של 30 מעלות. כשהפולימראז מבצע העתקה של גדיל התבנית ומייצר גדיל חדש, נוצרים מספר עותקים כאשר העותקים שנוצרו במחזור הקודם מוצאים החוצה מהתבנית. התוצרים של ריאקציית MDS הן באורך של כ-12 אלף נוקליאוטידים ויכולים להגיע לאורך של 100 אלף כך שיוכלו לשמש בריצוף דנ"א. שיטות אחרות המשמשות להגברה MALBAC.
מגבלות לשיטות ההגברה
לשיטת MDA יש הטיות בתהליך ההגברה בהשוואה לשיטת PCR הכוללות ייצוג-יתר או ייצוג חסר של חלקים מגדיל התבנית. כתוצאה מכך רצפים מסוימים אובדים ואותה הטיה נגררת למחזורי ההגברה הבאים. שתי שיטות להתגבר על ההטיות הן איגום של ריאקציות תא-בודד השייכות לתאים מאותו הסוג ואיגום לפני ביצוע הריאקציה. השיטות הנפוצות לזיהוי תאים מאותו סוג כוללות FISH ו-conformation characteristics.
רב צורניות של נוקלאוטיד בודד, סניפ SNPs, המהווה חלק משמעותי מהשונות הגנטית בגנום האנושי ושונות במספר העותקים CNV מהווים בעיה בריצוף ברמת התא הבודד. כך גם הכמות המזערית של חומר גנטי הנמצאת בתא בודד. לכן למרות ההגברה קשה לנתח באופן מדויק את כמויות הדנ"א מתא בודד משום שהכיסוי נמוך ורגיש לשגיאות. להגברה בשיטת ריבוי העתקות MDS הכיסוי הממוצע לגנום הוא 80% וסניפים שאינם מכוסים על ידי קריאות ייעלמו. בנוסף, בשיטה זו יש אחוז גבוה של השמטת אללים כלומר היעדר זיהוי של אללים מדוגמאות הטרוזיגוטיות. מספר אלגוריתמים לזיהוי סניפים נמצאים בשימוש אך אף אחד מהם אינו ספציפי לריצוף ברמת התא הבודד. בהגברה בשיטת ריבוי העתקות והשוניות במספר העותקים CNV קיימת בעיה של זיהוי שגוי של CNVs, ניתן להתגבר על בעיה זו באמצעות אלגוריתמים לזיהוי והורדת ה"רעש" והפקה נכונה של variants.
יישומים
המיקרוביוטה היא מטרה עיקרית של ריצוף בתא בודד בשל הקושי לגדל את הדוגמאות בתרבית. ריצוף ברמת התא הבודד היא אחת הדרכים לזיהוי של המיקרוביוטה והגנום שלה. המיקרואורגניזם הראשון שהיה בשימוש בטכנולוגיות ריצוף תא בודד היה החיידק. לכשיושלם תהליך המחקר ואיסוף הנתונים של אורגניזמים אלו יתגלו תפקידים חדשים אשר ישליכו על בריאות האדם[2].
ריצוף סרטן הוא אחד היישומים המתפתחים של ריצוף ברמת התא הבודד. ניתוח וקטגוריזציה של רקמת גידול סרטנית ברמת, מציאת ארגון מחדש של קטעי דנ"א, שונות ברמת נוקלאוטיד בודד SNV, וריצוף גנום מלא ברמת תא בודד[3]. scDNAseq - ריצוף דנ"א ברמת תא בודד שימושי במיוחד למחקר מוטציות של מולקולות ביולוגיות שהן מטרות לתרופות כגון קולטנים לטירוזין קינאז (הגנים EGFR ,PDGFRA וכו') כאשר השיטות הרגילות של ניתוח ברמת הרקמה כולה עלולות לפספס מוטציות ברמת התא הבודד אשר עלולות לאפשר יתרות במסלולים ביולוגיים וכך ליצור עמידות של הגידול הסרטני.
ריצוף ברמת תא בודד של ה-Methylome

ריצוף ברמת התא הבודד לכימות המתילציה של הדנ"א. השיטה דומה לריצוף דנ"א ברמת התא הבודד עם תוספת טיפול בביסולפיט לפני הריצוף. השיטות שבשימוש הן whole genome bisulfite sequencing,[5] and reduced representation bisulfite sequencing[6][7]

ריצוף רנ"א ברמת התא הבודד scRNA-seq
השיטות הנוכחיות לכימות המצב המולקולארי של התאים (פרופיל ביטוי הגנים דרך ריכוזי רנ"א-שליח) בין אם במערך-דנ"א (microarray) או RNA-seq מבצעות מיצוע של הסיגנאל על פני אלפי או מיליוני תאים. בהינתן השונות של אוכלוסיית תאים שיטות אלו מתעלמות מאינטראקציו בין תאים ומהשונות באוכלוסייה ההכרחית לשמירת תפקוד תקין של הרקמה ולהתפתחות של מחלות. לכן מיצוע של תאים מספק מידע חלקי בלבד על המצב המולקולארי של המערכת.[8]
ריצוף רנ"א ברמת התא הבודד מספק פרופיל ביטוי גנים של תאים בודדים. ניתן לזהות אוכלוסיות נדירות של סוגי תאים באמצעות ניתוח אשכולות ובכך לאפשר איפיון וזיהוי תתי-אוכלוסיות בתוך אוכלוסייה הטרוגנית. שונות באוכלוסייה של תאים סרטניים ניתן לייחס למוטציות נרכשות אך גם תאים הזהים גנטית ובאותה סביבה מראים שונות בפרופיל התבטאות הגנים והחלבונים שלהם [9]. בנוסף, רנ"א במספר עותקים נמוך אשר יכול להפעיל פונקציות חשובות בתא הוא בדרך כלל קשה לאיתור או נחשב לרעש בשיטות המסורתיות למיצוע מספר תאים. ריצוף ברמת התא הבודד של מספר תאים רב יכול לזהות רנ"א לא-נפוץ שכזה ולגלות את הפיזור במספר העותקים באוכלוסייה. הידע לגבי צורת התפלגות הביטוי יכול לשמש להבנת מנגנוני בקרת השעתוק לדוגמה בשיטות של Perturb-seq.[10]
תהליכים נסיוניים
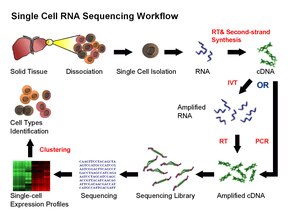
למרות ההתקדמות בטכנולוגיות הריצוף, עדיין לא ניתן לרצף רנ"א באופן ישיר מתאים בודדים. לכן בפרוטוקולי scRNA-seq הקיימים נדרש להמיר את רנ"א ל-דנ"א-משלים (cDNA) לצורכי ריצוף. השיטות הקיימות ל- scRNA-seq כוללות את השלבים הבאים: הפרדה של הרקמה לתאים בודדים ומיצוי רנ"א מכל תא, שיעתוק-הפוך (reverse transcription), הגברה, הכנת ספרייה וריצוף.
באופן מושלם, scRNA-seq אמור לשמר ולכמת את השכיחות היחסית של מולקולות רנ"א-שליח בתא, מכסה באופן מלא לכל אורך התעתיק ובייצוג שווה לכל נקודה ושומר על מידע לגבי הגדיל. למרות זאת, בשלבים של scRNA-seq נכנסים מספר הטיות ורעשים. לדוגמה, התהליך של שעתוק הפוך הוא קריטי שכן יעילות של השעתוק ההפוך קובעת את אחוז הרנ"א בתא שיעבור ריצוף בסופו של דבר. היעילות של האנזים המבצע את תהליך השעתוק ההפוך (reverse transcriptase) ושל שיטת הצמדת ה-תחל (primer) ישפיע על היווצרות בדנ"א המשלים (cDNA) ועל יצירת הספריות והטיה שלהן לכיוון '3 או '5 של הגן. בשלב ההגברה משתמשים ב-PCR או In Vitro Transcription להגברת דנ"א-משלים (cDNA). אחת היתרונות של שיטות מבוססות PCR היא היכולת ליצור דנ"א-משלים באורך מלא. בנוסף, יעילות התהליך ה-PCR משתנה ברצפים מסוימים לדוגמה רצפים עשירים בנוקלאוטידים מסוג GC מה שמייצר ספריות עם כיסוי לא אחיד.
מצד שני בספריות שנוצרו על ידי In Vitro Transcription אפשר להימנע מהטיות כפי שנוצרות על ידי PCR אולם רצפים מסוימים ישועתקו ביעילות פחותה וכך ייצרו רצפים שאינם שלימים פורסמו מספר שיטות של scRNA-seq: Tang et al.[11], STRT[12], SMART-seq[13], CEL-seq[14] and Quartz-seq[15].
יישומים
הוכח כי יש קשר בין תאים סרטניים המתגלים בדם של חולי סרטן לבין הפרוגנוזה של החולה[16]. . אולם קשה לכמת ולאפיין את בין התאים האלו במחזור הדם שכן הם מזוהמים בתאי דם לבנים ואדומים. ריצוף רנ"א בתאים בודדים יכול לשמש לבידול תאים סרטנים מתאי דם וכן על מנת לקבל פרופיל ביטוי גנים של תאי הגידול. בדומה לכך ניתן להשתמש ב-scRNA-seq לנתח אוכלוסייה נדירה של תאים עובריים בשלבים מוקדמים וכן אוכלוסייה של תאי אב בוגרים, שני סוגי תאים הקיימים באופן ארעי. לבסוף ניתן להשתמש בניתוח של תאים בודדים למחקר מחלות מידבקות[17].
שיקולים נוספים
Isolation of single cells
אין כעת שיטה סטנדרטית לבידוד של תאים בודדים. ניתן לאסוף תאים בודדים באמצעות שיטות של מיקרומניפולציה למשל על ידי דילול מספר פעמים או על ידי שימוש ב-פִּיפִית (פיפטה)[18][19]. היתרון של שיטות מיקרומניפולציה הוא שהן זולות אך הן מייגעות ורגישות לטעויות בזיהוי סוג התא תחת מיקרוסקופ. שיטות של לכידה באמצעות מיקרו-חיתוך לייזר (LCM - Laser Capture Microdissection) יכולות לשמש לאיסוף תאים בודדים. בשיטה זו למרות שהלייזר שומר את המידע המרחבי לגבי מיקום התא, קשה לאסוף תא בודד מבלי לאסוף חומר מהתאים שמסביבו .[20][21][22] שיטות בתפוקה גבוהה להפקה של תאים בודדים כוללות (fluorescence-activated cell sorting (FACS) ושיטות מיקרופלואידיות. גם FACS וגם שיטות מיקרופלואידיות הן מדויקות ואוטומטיות ואינן מוטות אולם שיטות אלו מחייבות הסרה של התאים ממיקרו-הסביבה ולכן גורמות לשינויים בפרופיל התעתיקי של ה-רנ"א[23][24].
מספר התאים לניתוח
scRNA-Seq
באופן כללי בניסוי RNA-seq מיוצרים כ-10 מיליון קריאות וגן עם מעל 50 קריאות לכל kb לכל מיליון קריאות (RPKM) נחשב גן שמתבטא. כלומר, עבור גן שאורכו 1kb המשמעות היא רמת ביטוי של 500 קריאות ומקדם שונות (Coefficient of Variation) של 4% לפחות תחת ההנחה של התפלגות פואסון. עבור תא מאורגניזם ששייך למחלקת היונקים המכיל 200,000 מולקולות רנ"א-שליח, נדרש לצרף נתונים לאחר הריצוף מכ- 50 תאים על מנת להשיק את מקדם השונות הזה. אולם בשל בעיות הנובעות מיעילות תהליך השיעתוק ההפוך ורעשים נוספים נדרשים יותר תאים על מנת לבצע ניתוח וזיהוי של סוגי תאים.
הערות שוליים
- ↑ "The promise of single-cell sequencing". Nat. Methods. 11 (1): 25–27. בינואר 2014. doi:10.1038/nmeth.2769. PMID 24524134.
{{cite journal}}: (עזרה) - ↑ Blainey, PC.; Quake SR. (2014). "Dissecting genomic diversity, one cell at a time". Nat Methods. 11 (1): 19–21. doi:10.1038/nmeth.2783. PMC 3947563. PMID 24524132.
- ↑ (Francis J, Zheng CZ, Maire C et al.
- ↑ Farlik, M; Sheffield, NC; Nuzzo, A; Datlinger, P; Schönegger, A; Klughammer, J; Bock, C (3 במרץ 2015). "Single-cell DNA methylome sequencing and bioinformatic inference of epigenomic cell-state dynamics". Cell reports. 10 (8): 1386–97. doi:10.1016/j.celrep.2015.02.001. PMC 4542311. PMID 25732828.
{{cite journal}}: (עזרה) - ↑ Smallwood, SA; Lee, HJ; Angermueller, C; Krueger, F; Saadeh, H; Peat, J; Andrews, SR; Stegle, O; Reik, W (באוגוסט 2014). "Single-cell genome-wide bisulfite sequencing for assessing epigenetic heterogeneity". Nature Methods. 11 (8): 817–20. doi:10.1038/nmeth.3035. PMC 4117646. PMID 25042786.
{{cite journal}}: (עזרה) - ↑ Guo, H; Zhu, P; Wu, X; Li, X; Wen, L; Tang, F (בדצמבר 2013). "Single-cell methylome landscapes of mouse embryonic stem cells and early embryos analyzed using reduced representation bisulfite sequencing". Genome Research. 23 (12): 2126–35. doi:10.1101/gr.161679.113. PMC 3847781. PMID 24179143.
{{cite journal}}: (עזרה) - ↑ Guo, H; Zhu, P; Guo, F; Li, X; Wu, X; Fan, X; Wen, L; Tang, F (במאי 2015). "Profiling DNA methylome landscapes of mammalian cells with single-cell reduced-representation bisulfite sequencing". Nature Protocols. 10 (5): 645–59. doi:10.1038/nprot.2015.039. PMID 25837417.
{{cite journal}}: (עזרה) - ↑ Kolodziejczyk, Aleksandra A.; Kim, Jong Kyoung; Svensson, Valentine; Marioni, John C.; Teichmann, Sarah A. (במאי 2015). "The Technology and Biology of Single-Cell RNA Sequencing". Molecular Cell. 58 (4): 610–620. doi:10.1016/j.molcel.2015.04.005.
{{cite journal}}: (עזרה) - ↑ "Using gene expression noise to understand gene regulation". Science. 336 (6078): 183–7. 2012. doi:10.1126/science.1216379. PMC 3358231. PMID 22499939.
- ↑ "Hebenstreit, D. (2012). "Methods, Challenges and Potentials of Single Cell RNA-seq". Biology. 1 (3): 658–667. doi:10.3390/biology1030658.
- ↑ Tang, F.; Barbacioru C; Wang Y; et al. (2009). "mRNA-Seq whole-transcriptome analysis of a single cell". Nat Methods. 6 (5): 377–82. doi:10.1038/NMETH.1315. PMID 19349980.
- ↑ Islam, S.; Kjällquist U; Moliner A; et al. (2011). "Characterization of the single-cell transcriptional landscape by highly multiplex RNA-seq". Genome Res. 21 (7): 1160–7. doi:10.1101/gr.110882.110. PMC 3129258. PMID 21543516.
- ↑ Ramsköld, D.; Luo S; Wang Y-C; et al. (2012). "Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells". Nat. Biotechnol. 30 (8): 777–82. doi:10.1038/nbt.2282. PMC 3467340. PMID 22820318.
- ↑ ,Hashimshony, T.; Wagner F; Sher N; Yanai I. (2012). "CEL-Seq: single-cell RNA-Seq by multiplexed linear amplification". Cell Rep. 2 (3): 666–73. doi:10.1016/j.celrep.2012.08.003. PMID 22939981.
- ↑ Sasagawa, Y.; Nikaido I; Hayashi T; et al. (2013). "Quartz-Seq: a highly reproducible and sensitive single-cell RNA sequencing method, reveals non-genetic gene-expression heterogeneity". Genome Biol. 14 (4): R31. doi:10.1186/gb-2013-14-4-r31. PMC 4054835. PMID 23594475.
- ↑ Olmos, D.; Arkenau H-T; Ang JE; et al. (2009). "Circulating tumour cell (CTC) counts as intermediate end points in castration-resistant prostate cancer (CRPC): a single-centre experience". Ann. Oncol. 20 (1): 27–33. doi:10.1093/annonc/mdn544. PMID 18695026.
- ↑ Avraham, Roi; Haseley, Nathan; Brown, Douglas; Penaranda, Cristina; Jijon, Humberto B.; Trombetta, John J.; Satija, Rahul; Shalek, Alex K.; Xavier, Ramnik J.; et al. (10 בספטמבר 2015). "Pathogen Cell-to-Cell Variability Drives Heterogeneity in Host Immune Responses". Cell. Elsevier BV. 162 (6): 1309–1321. doi:10.1016/j.cell.2015.08.027. ISSN 0092-8674. PMC 4578813. PMID 26343579.
{{cite journal}}: (עזרה) - ↑ Zong, C; Lu S; Chapman AR; Xie XS. (2012). "Genome-wide detection of single-nucleotide and copy-number variations of a single human cell". Science. 338 (6114): 1622–6. doi:10.1126/science.1229164. PMC 3600412. PMID 23258894.
- ↑ Kurimoto, K.; Yabuta Y; Ohinata Y; Saitou M. (2007). "Global single-cell cDNA amplification to provide a template for representative high-density oligonucleotide microarray analysis". Nat Protoc. 2 (3): 739–52. doi:10.1038/nprot.2007.79. PMC 3600412. PMID 17406636.
- ↑ "Shapiro, E.; Biezuner T; Linnarsson S.; Saitou M. (2013). "Single-cell sequencing-based technologies will revolutionize whole-organism science". Nat Rev Genet. 14 (9): 618–30. doi:10.1038/nrg3542. PMID 23897237.
- ↑ Yachida, S.; Jones S; Bozic I; et al. (2010). "Distant metastasis occurs late during the genetic evolution of pancreatic cancer". Nature. 467 (7319): 1114–7. doi:10.1038/nature09515. PMC 3148940. PMID 20981102.
- ↑ Frumkin, D.; Wasserstrom A; Itzkovitz S; Harmelin A; Rechavi G; Shapiro E.; et al. (2008). "Amplification of multiple genomic loci from single cells isolated by laser micro-dissection of tissues". BMC Biotechnol. 8 (17): 1114–7. doi:10.1186/1472-6750-8-17. PMC 2266725. PMID 18284708.
- ↑ Dalerba, P.; Kalisky T; Sahoo D; et al. (2011). "Single-cell dissection of transcriptional heterogeneity in human colon tumors". Nat. Biotechnol. 29 (12): 1120–7. doi:10.1038/nbt.2038. PMC 3237928. PMID 22081019.
- ↑ White, AK.; VanInsberghe M; Petriv OI; et al. (2011). "High-throughput microfluidic single-cell RT-qPCR". Proc Natl Acad Sci U S A. 108 (34): 13999–4004. doi:10.1073/pnas.1019446108. PMC 3161570. PMID 21808033.

שגיאות פרמטריות בתבנית:מיון ויקיפדיה
שימוש בפרמטרים מיושנים [ דרגה ] ריצוף ברמת התא הבודד24833251